
The structure, function, and dynamics of proteins are dictated by the molecular interaction between its residues and the interaction with its environment. Some of the most important interactions are hydrogen bond, electrostatic, and non polar interations. And it is important to note that for some residues these interactions were affected by their protonation states. For example the deprotonation of aspartate, glutamate, and cysteine residue will cause them unable to act as hydrogen donor. Also, the protonation or deprotonation of a residue can cause formal charge change, which could modulate the short range and long range electrostatic interactions. Thus, in order to properly model and simulate a protein it is important to calculate the protonation state of the titratable residues in the protein.
In this post I would like to talk about the details regarding the protonation state of several titratable residues and how it could affect the structure, function and dynamics of a protein. Then, I would continue with some of the use case where protonation state determination is important in protein-ligand modeling and protein simulations. Lastly, I would like to discuss some of the methods that can be used in determining the protonation states of titratable residues.
Titratable residues and its protonation states variant
Out of 20 common amino acid residues, seven of them are titratable. Which can be categorized to three classes:
- Carboxylic acid residues: aspartic acid and glutamic acid. These residues contain carboxylic acid which is usually negatively charged under physiological pH and may turn neutral under pH 4-5.
- Ammonium and Guanidinium residues: lysine and arginine. These residues contain basic moiety that have high pK, therefore it is rare to find lysine in neutral state at physiological pH, and even more rare to find neutral arginine even at high pH like 11 or 12. Usually arginine is always treated as positively charged as its pK usually ranging from 12 to 14.
- Special case residues: cysteine, tyrosine, histidine. Cysteine and tyrosine are basically weakly acidic residues and usually neutral at physiological pH. Cysteine have lower pK than tyrosine and thus although very rare, in some condition it can be deprotonated around physiological pH. Histidine on the other hand is a weakly basic residue, although most of the time the imidazole moiety is neutral under physiological pH, both of the nitrogen atoms in the imidazole ring could be protonated under the right condition.
Things that could affect the protonation state of titratable residues
There are many factors that could affect the protonation states of titratable residues, but basically they all come down to the microenvironment surrounding the residues, things like the hydrogen bond network, solvent accessibility AKA residue burriedness, and the electrostatic charge.
For example, the existence of a proton donor next to carboxylic acid residues might form hydrogen bond and stabilize the negatively charged form of these residues, and thus lowering their pK. In the same way, when charged residue have access to water then the charged form will be stabilized, on the opposite, when it is burried it will be more stable in its neutral form.
The electrostatic charge surrounding the titratable residues will depend on the charge. If the electrostatic charge surrounding the residue is the opposite charge then the charged residue will be stabilized and vice versa.
Various Method for determining the protonation state of titratable residues
To this date, there are four different computational-based approach to predict or determine the protonation states of titratable residues. I would start from the first-principle approach, then continuing to the empirical approach which includes MD and electrostatic-based method, and lastly the most recent approach, the machine learning method.
QM-MM method
One of the most accurate way to determine the protonation states of titratable residues in protein is using QM/MM. Ideally, quantum mechanics can be used to do this, but since protein is very large, doing QM calculation for the whole system is computationally expensive and thus not practical. To tackle this issue, QM can be combined with molecular mechanics (MM) by applying the QM to the residue of interest (i.e. catalytic site, ion channel pore) and its surrounding, then, the rest of the protein is treated with MM.
Then, multiple protonation state configuration can be simulated with QM/MM to find the relative stability for each configuration. The more titratable residue tested inside QM environment the more configuration that should be simulated, usually the number of configuration is 2n where n is the number of the titratable residues. For example, if the catalytic site being simulated contain Asp213, Asp335, and Lys370 then there should be 8 different configuration that must be simulated as shown in the following table:
| config num | Asp213 | Asp335 | Lys370 |
|---|---|---|---|
| 1 | protonated | protonated | protonated |
| 2 | protonated | protonated | neutral |
| 3 | protonated | neutral | protonated |
| 4 | protonated | neutral | neutral |
| 5 | neutral | protonated | protonated |
| 6 | neutral | protonated | neutral |
| 7 | neutral | neutral | protonated |
| 8 | neutral | neutral | neutral |
The most stable protonation state for each residues can be derived from the resulting relative stability measured from QM/MM simulation.
Constant pH Molecular Dynamics
This method is similar to the QM/MM simulation, but rather than using QM this method uses full MM for the simulation. Also, the protonation states are allowed to change over time. This is possible by implementing nonequilibrium MD and integrating Monte Carlo so that the protonotation state switch will be accepted if it pass the Metropolis criterion otherwise it will be rejected.
Electrostatic-based method
This method predict the protonation states of titratable residues by calculating the pK values using electrostatic interactions. These are done by calculating the the microenvironment surrounding the titratable residues that might affects the pK value such as the neighboring residues, solvent, ions.
After the pK value is retrieved the protonation states can be predicted by measuring the differences between the pK and the intended pH. It is common to use 1.0 unit as the ptreshold value, for example if the aspartate residue has the pK 5.7 then the aspartate would be considered neutral at pH 6 because the pH-pK difference is only 0.3, but it would be considered as negatively charged at ph 7 because the pH-pK difference is larger than 1.0. The value 1.0 is chosen because according to Henderson-Hasselbach equation (see below) when the difference between pH and pK is 1.0 then the concentration ratio between the neutral state and the ionized state is 1 to 10. Which is good enough to make sure that most of the titratable residue is ionized.
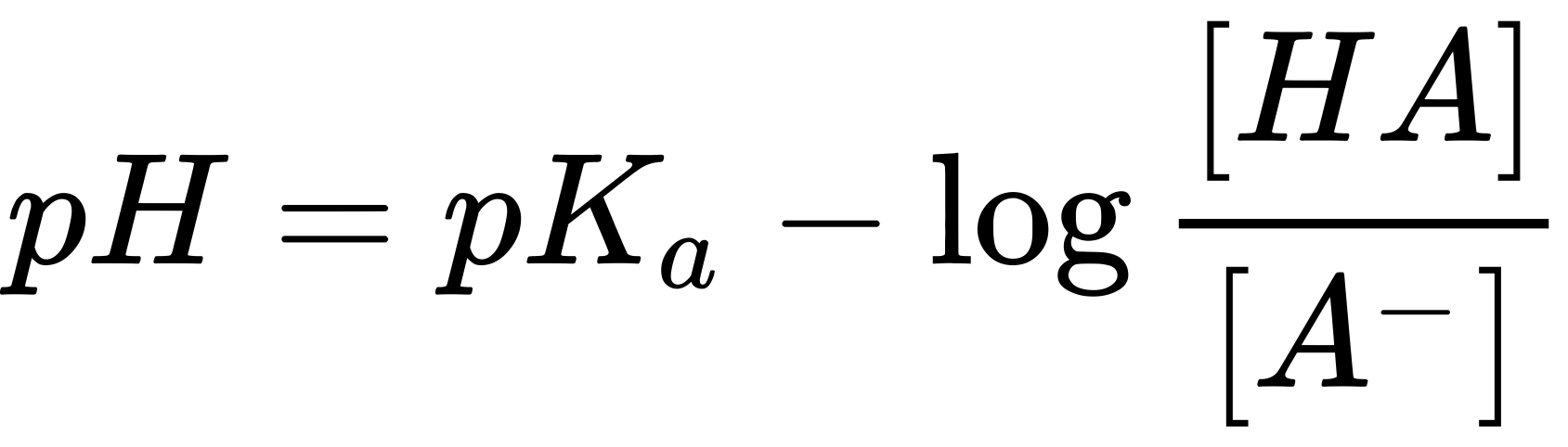
To this date, this is the most common method and give good balance between accuracy and speed. There are many tools that use this method such as Delphi, PROPKA, PypKa, and H++.
Machine-learning method
In this decade there are many novel computational chemistry methods that utilize machine-learning to enhance the accuracy and the efficiency of the calculation. Starting from the use of graph neural network for modeling protein-ligand interaction, ANI-2x for QM calculation, to machine-learning based forcefield. Recently, new work that utilizes deep learning for the pK prediction of titratable residues has been published by Reis et al. This method is implemented as command line tool pKAI which has been shown to be comparable with electrostatic-based method such as PROPKA.
pKAI works by using deep learning to learn the relationship between the pK values of titratable residues and its surrounding, and generate the model that could predict the pK based on the residue name and its surrounding. Like the previous method, the predicted pK can be used to determine the protonation state of the residue.
Concluding remark
Protonation state determination is a crucial step in protein preparation prior to molecular simulation as it may affects how it binds with other molecule, the stability of the protein structure, and how realistic and how close it resembles the protein structure and behavior in nature.
There are several things that need to be considered in determining the protonation state of titratable residues. First, do we have to predict accurately the protonation state of few residue of interest, or we need to determine all of the protonation state at acceptable accuracy. If it is the former case, like in the simulation of catalytic site or ion transportation in a protein then it would be best to use very accurate method but focused on small portion of the protein like QM/MM or in some case CpHMD could give reasonable accuracy for this. On the other hand, if it is the latter case, than electrostatic-based method or ML-based method should do. To compare which one is best for your research it is best to learn from their benchmarking.